Bioinformática - PhyML: alinhamento de sequências nucleotídicas em ambiente paralelo
O PhyML é um software utilizado para se fazer estimativa e probabilidade filogênica em alinhamento de sequências nucleotídicas ou sequências de anino ácido. Atualmente está na versão 3.0, seu desenvolvimento teve participação de diversos cientistas de algumas universidades no mundo.
[ Hits: 32.073 ]
Por: José Cleydson Ferreira da Silva em 27/07/2010

Sumário - Sobre o PhyML
Sumário
1. Sobre o PhyML2. Menus do PhyML
2.1 Submenu de entrada de dados
2.2 Submenu de modelos de substituição
2.3 submenu - Pesquisa de Árvore
3. Usando PhyML em linha de comando
4. Instalando PHYML em ambiente paralelizado
4.1Download do PHYML
5.0 Sobre o autor
6.0 Referências
1. Sobre o PhyML
O PhyML é um software utilizado para se fazer estimativa e probabilidade filogênica em alinhamento de sequências nucleotídicas ou sequências de anino acido. Atualmente está na versão 3.0, seu desenvolvimento teve participação de diversos cientistas de algumas universidades como apresenta o quadro abaixo: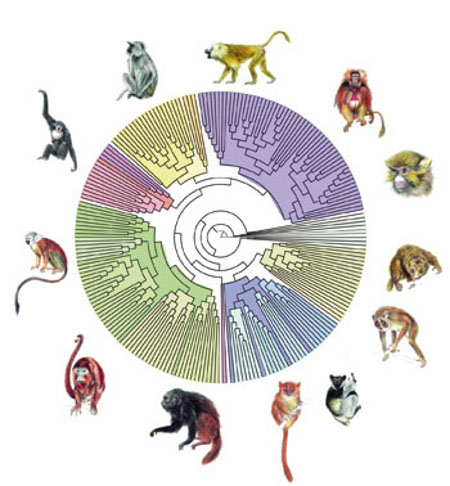
Departamento de Estatística. Universidade de Auckland. New Zealand
Universidade de Montpellier II - CNRS UMR 5506. LIRMM. Montpellier França.
e-mail : guindon@stat.auckland.ac.nz
Olivier Gascuel
Universidade Montpellier II - CNRS UMR 5506. LIRMM. Montpellier França.
e-mail : gascuel@lirmm.fr
Jean-Francois Dufayard
Universidade Montpellier II - CNRS UMR 5506. LIRMM. Montpellier França.
e-mail : jeanfrancois.dufayard@gmail.com
** Authors (before 2006)... **
Wim Hodrijk
Atualmente trabalhando na 'Rede nacional de Bioinformática - National Bioinformatics Network'. Cape Town, Africa do Sul
e-mail : wim@santafe.edu
Franck Lethiec
Universidade Montpellier II - CNRS UMR 5506. LIRMM. Montpellier França.
e-mail : lethiec@lirmm.fr
Este software é utilizado em linha de comando e possui uma interface simples com diversas diretivas de configuração e parâmetros que correspondem métodos estáticos, analise de sequência e entre outros.
Os métodos utilizado pelo PhyML dá-se também por Máxima Verosimilhança para reconstrução Filogenética, o que possibilita encontrar a árvore que mais provavelmente explicaria a evolução de determinado indivíduo . Em seguida podemos verificar os menus e suas funções.
2. Menus do PhyML
Os parâmetros de configuração podem ser utilizados de duas formas: Um por meio da interface do software e outro por meio de comandos shell. A interface do pode ser usada acessando menus conforme as letras que identificam as funções, a seguir podemos apresentá-las.2.1 Submenu de entrada de dados
[D] ............................... Data type (DNA/AA)Tipo de dados. Podendo ser DNA ou aminoácido.
[I] ...... Input sequences interleaved (or sequential)
Entrada de intercalada de sequência.
[M] ....................... Analyze multiple data sets
Analise múltipla de dados.
2.2 Submenu de modelos de substituição
[M] ................. Model of nucleotide substitutionModelo de substituição de Nucleotídios
[M] ................ Model of amino-acids substitution
Modelo de substituição de aminoácidos
[F] ................. Optimise equilibrium frequencies
Otimizar frequência
[F] . Amino acid frequencies (empirical/model defined)
Frequência de aminoácidos com modelo empírico definido
[T] .................... Ts/tv ratio (fixed/estimated)
Fixar ou estimar transcrição / transversão com relação a Máxima Verosimilhança
[V] . Proportion of invariable sites (fixed/estimated)
Proporção de viabilidade fixa ou estimada
[R] ....... One category of substitution rate (yes/no)
Categoria de substituição de taxa
[C] ........... Number of substitution rate categories
Numero de substituição da categoria taxas
2.3 submenu - Pesquisa de Árvore
[O] ........................... Optimise tree topologyOtimizar topologia da árvore
[S] .................. Tree topology search operations
Operações de pesquisa na topologia da árvore
[R] ......................... Use random starting tree
Iniciar árvore usando aleatoriedade
[N] .................. Number of random starting trees
Iniciar árvore com numero aleatório
[U] ..................... Input tree (BioNJ/user tree)
Entrar com uma árvore
2.4 Suporte a Brunch
[B] ................ Non parametric bootstrap analysisSem analise para métrica bootstrap
[A] ................ Approximate likelihood ratio test
Teste de aproximação Verosimilhança.
2. Usando PhyML em linha de comando
3. Instalando PHYML em ambiente paralelizado
4. Sobre autor
Bioinformática - Análise Filogenética com Clustalx
Implementando servidor de aplicações PHP utilizando Zend Framework
Compiz - Janelas à 360 graus no Linux
FreePascal + Lazarus: Desenvolvedores em Delphi podem começar a migrar para o Linux!
Automação e Sensoreamento Remoto utilizando Software Livre "SCADA"
ERPs Open Source (parte 1) - Principais soluções
Experiência no desenvolvimento de software para automação comercial
Grande Kuruma!!
Parabéns pelo artigo. Apesar de eu não estar familiarizado com coisas como:
probabilidade filogênica
alinhamento de sequências nucleotídicas
sequências de anino ácido
Eu acho que isso irá e muito ajudar a comunidade do software livre.
E mais do que nunca
VIDA LONGA AO SL!!
Dailson Fernandes
http://www.dailson.com.br
Olá Dailson,
Grande Guerreiro, obrigado pelo incentivo meu camarada. Vou começar uma serie longa desses artigos, estou pensando em enviar um trabalho para o VOL DAY, em Bebedouro-SP, mas estou pensando se poderá ser aceito ou não. Esses artigos com certeza deram muito trabalho para validar a prática. Mas com certeza resolverá a vida dos Biologos e dos Bioinformatas.
Um abraço!
Patrocínio

Destaques
Artigos
Maquina modesta - a vez dos navegadores ferrarem o usuario
Fscrypt: protegendo arquivos do seu usuário sem a lentidão padrão de criptograr o disco
Faça suas próprias atualizações de pacotes/programas no Void Linux e torne-se um Contribuidor
Como rodar o Folding@home no Linux
Criando um painel de controle (Dashboard) para seu servidor com o Homepage
Dicas
Configurando o Cairo Dock individualmente em ambientes diferentes na mesma maquina
Calculadoras online gratuitas para o dia a dia do usuário Linux
Utilizando a Ferramenta xcheckrestart no Void Linux
Pisando no acelerador do Linux Mint: Kernel XanMod, zRAM e Ajustes de Swap
Tópicos
Warsaw não é reconhecido no Google Chrome 147.0.7727.55 (1)
Meu kde connect não funciona no debian (1)
Top 10 do mês
-

Xerxes
1° lugar - 150.137 pts -

Fábio Berbert de Paula
2° lugar - 71.190 pts -

Buckminster
3° lugar - 49.122 pts -
Alberto Federman Neto.
4° lugar - 39.810 pts -

edps
5° lugar - 25.774 pts -

Alessandro de Oliveira Faria (A.K.A. CABELO)
6° lugar - 25.655 pts -

Sidnei Serra
7° lugar - 24.690 pts -

Mauricio Ferrari (LinuxProativo)
8° lugar - 22.540 pts -

Daniel Lara Souza
9° lugar - 22.310 pts -

Andre (pinduvoz)
10° lugar - 18.682 pts
Scripts
A maior comunidade GNU/Linux da América Latina! Artigos, dicas, tutoriais, fórum, scripts e muito mais. Ideal para quem busca auto-ajuda.
Site hospedado por: