Bioinformática - Análise Filogenética com Clustalx
O Clustalx é um software para o alinhamento de sequências nucleotídicas, peptídicas e analises filogênicas. O Clustalx é software semi-livre segundo a sua forma de licenciamento. Embora possua características de software livre como permissão de execução de programa, não há permissão de uso para fins comerciais.
[ Hits: 29.418 ]
Por: José Cleydson Ferreira da Silva em 08/07/2010

Usando o Clustalx

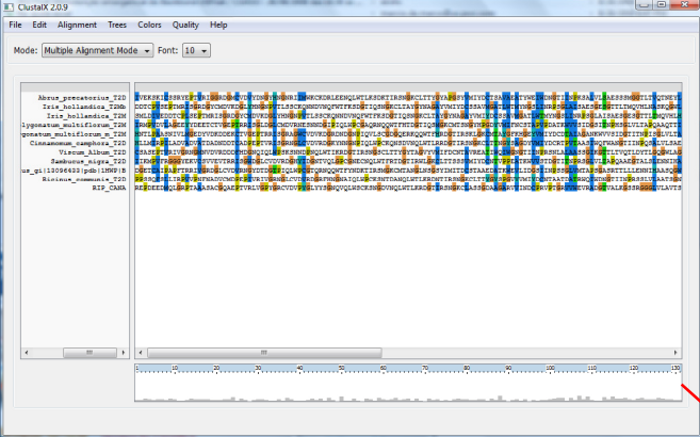


- Gap Opening- penalização de escore para iniciar uma região de "Gap".
- Gap Extension- penalização de escore para estender uma região de "Gap" (normalmente menor que o Gap Opening)
- Delay Divergent Sequences - atraso de alinhamento de sequências divergentes que somente serão alinhadas após as outras sequências (porcentagem de identidade abaixo da qual a sequência será considerada divergente)
- Transition Weight (somente DNA) - da a transições (A<->G, T<->C) um escore diferente de 0.
- Use negative matrix - permite o uso de matrizes negativas, importante quando as sequências forem relacionadas somente em uma pequena porção. Em condições normais prejudica um pouco o alinhamento.
- Protein Weight Matrix - matriz de substituição a ser utilizada. Note que você só escolhe a serie de matriz: Blosum, PAM etc, o tipo de matriz dentro desta série (por exemplo Blosum 62, blosum 80) é escolhido automaticamente pelo programa.
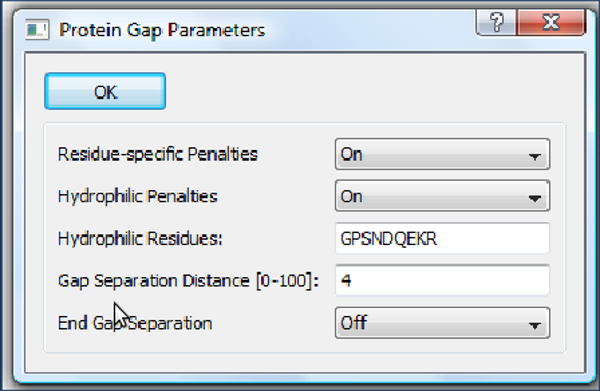
- Residue-specific Penalties - considera a vizinhança de alguns resíduos como mais ou menos favorável para a abertura de "Gaps".
- Hydrophilic Penalties - aumenta a chance de Gaps em regiões ricas em resíduos hidrofílicos que, usualmente, representam regiões menos estruturadas.
- Hydrophilic Residues - especifica quais resíduos são considerados hidrofílicos.
- Gap Separation Distance - número de resíduos de distância de uma região com Gap na qual é penalizada uma nova abertura de Gap.
A apresentação de resultado de um alinhamento no ClustalX é representada por símbolos que indicam a conservação e os resíduos ou grupos de resíduos em uma coluna.
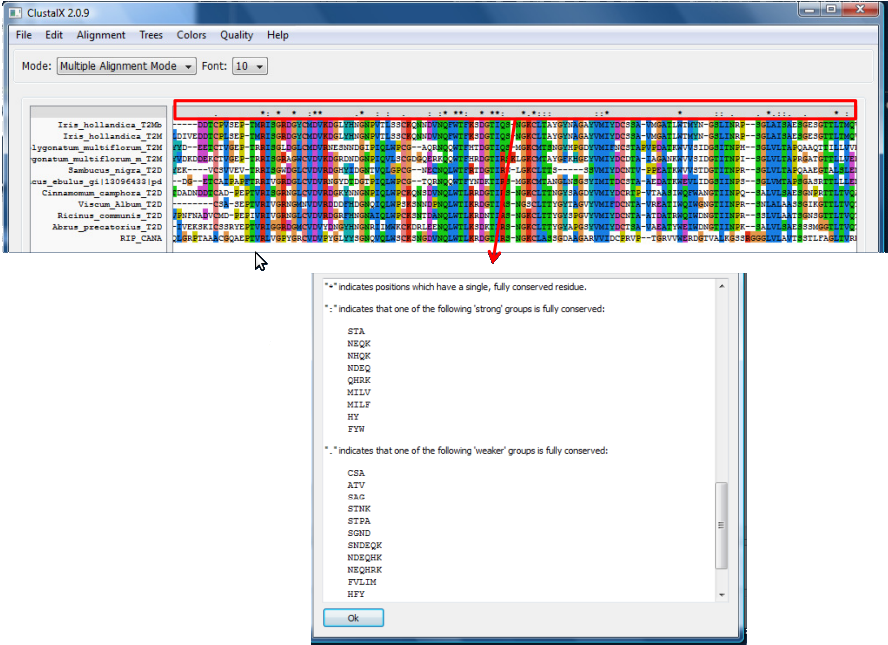
- Low Scoring Segments - mostra, em cinza, regiões com baixo escore e que, portanto, não seriam muito confiáveis
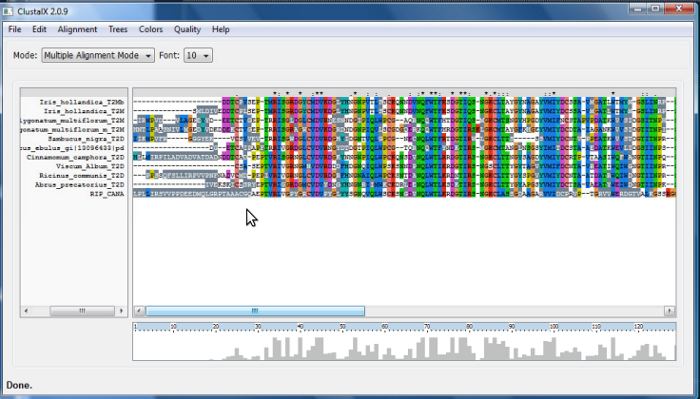
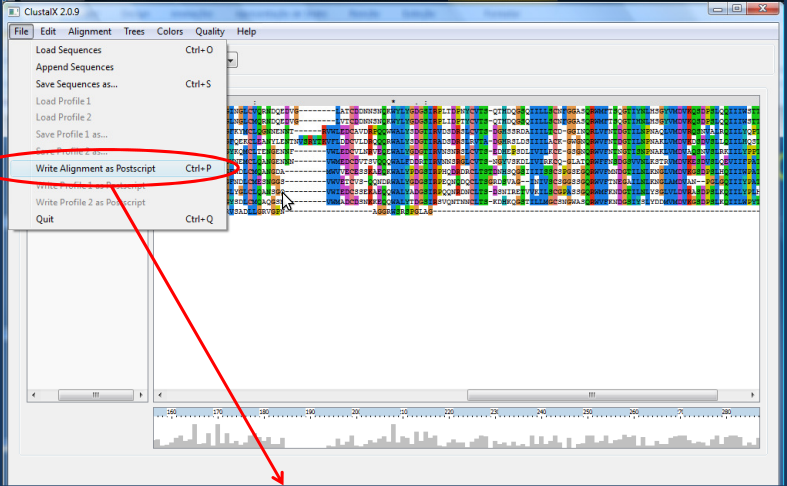
2. Usando o Clustalx
3. Sobre o autor
Bioinformática - Instalação do Mr Bayes em ambiente paralelo
Bioinformática - PhyML: alinhamento de sequências nucleotídicas em ambiente paralelo
Economia e liberdade: o software livre une o útil ao agradável
Projetos de software livre descontinuados: um problema com solução
Acessibilidade: Movimentos do mouse com a face (eViacam)
S1 Ponto: Sistema de controle de ponto Open Source para Linux (Ubuntu)
Baixando ISOs-Linux em altíssima velocidade
Nenhum comentário foi encontrado.
Patrocínio

Destaques
Artigos
Como rodar o Folding@home no Linux
Criando um painel de controle (Dashboard) para seu servidor com o Homepage
O Abismo entre o Código e o Chão: Saltos Tecnológicos e a Exclusão Estrutural no Brasil
Instalar e Configurar a santíssima trindade (PAP) no Void Linux
Dicas
Lançamento do Brutal DOOM test 6
Consertando o erro no Brave de webgl
Solução para ter de volta as bordas e barra de títulos das janelas em zenity no Debian 13.x
NixOS + NVIDIA antiga: como sobreviver ao driver 595 (GTX 750 Ti / Maxwell)
Tópicos
O que você está ouvindo agora? [2] (244)
Qual melhor distro para esse notebook? (1)
Top 10 do mês
-

Xerxes
1° lugar - 139.553 pts -

Fábio Berbert de Paula
2° lugar - 64.686 pts -

Buckminster
3° lugar - 43.646 pts -
Alberto Federman Neto.
4° lugar - 35.492 pts -

Sidnei Serra
5° lugar - 24.334 pts -

Alessandro de Oliveira Faria (A.K.A. CABELO)
6° lugar - 22.914 pts -

edps
7° lugar - 22.633 pts -

Daniel Lara Souza
8° lugar - 20.413 pts -

Mauricio Ferrari (LinuxProativo)
9° lugar - 19.981 pts -

Andre (pinduvoz)
10° lugar - 16.817 pts
Scripts
A maior comunidade GNU/Linux da América Latina! Artigos, dicas, tutoriais, fórum, scripts e muito mais. Ideal para quem busca auto-ajuda.
Site hospedado por: